随着circRNA研究的持续深入,人们不断发现在circRNA中也偶尔会存在于母基因序列不一致的情况,因此circRNA全长序列的鉴定和分析变得越来越重要。但如何才能分析和得到circRNA的全长序列呢?有没有方便的工具能直接实现这个目标?2月3日,Genomics, Proteomics & Bioinformatics杂志(IF=6.597)在线发表了南京航空航天大学宋晓峰教授团队的一项研究成果,报道一种可以分析circRNA全长的生信工具:circAST([1])。下面就让我们一起来了解一下这个工具的主要原理和使用情况:

circRNA与mRNA一样是从转录产物剪切形成,只是circRNA中存在下游与上游结合的反向剪切方式(BSJ),并因此形成环形序列。circRNA很多时候与mRNA的外显子是一致的,但也存在circRNA特有的剪切形式。那么本文中作者开发的circAST是如何基于转录组测序的片段化序列拼接出circRNA全长信息的?如何实现circRNA产物的定量?circAST分析circRNA的效果怎么样?与其他circRNA分析工具相比如何?
circAST是如何分析circRNA全长的?
circAST拼接circRNA全长的算法概括而言,是基于构建多重的RNA拼接图模型,从对应的转录组测序片段和基因组比对信息中拼接出全长circRNA信息,最终的找到能够解释所有测序片段的最少circRNA拼接形式组合。具体的做法如下:首先基于Tophat2将所有测序片段中携带了拼接位点的Reads分析出来,即找出确定是跳过一段基因组序列(内含子)的那些片段,这里面包含了对应的外显子边界信息。这些测序片段通过之前报道的circRNA分析工具分析出其中的反向拼接方式,构建出RNA反向拼接图(splice graphs)。传统的线性mRNA转录产物拼接算法一般对每个基因只给定一种转录拼接图(splice graphs),但circAST将每个反向拼接的产物都作为独立的splice graphs,并基于这个进行序列拼接,因此可以有效的将反向拼接事件进行准确拼接组装。
circAST将circRNA拼接过程认为是一个扩展的最小路径覆盖问题(extended minimum path cover,EMPC),该算法通过分析测序产物中最优化的拼接方式,从BSJ的两侧均分析可能的拼接路径,最终实现circRNA全长的拼接组装。(具体算法原理详见原文,如果有理解错误敬请批评指正)。
为有效的计算各种反向拼接产物的丰度,circAST用期望最大化算法(expectation maximization algorithm,EM)对各种剪切方式进行丰度计算。
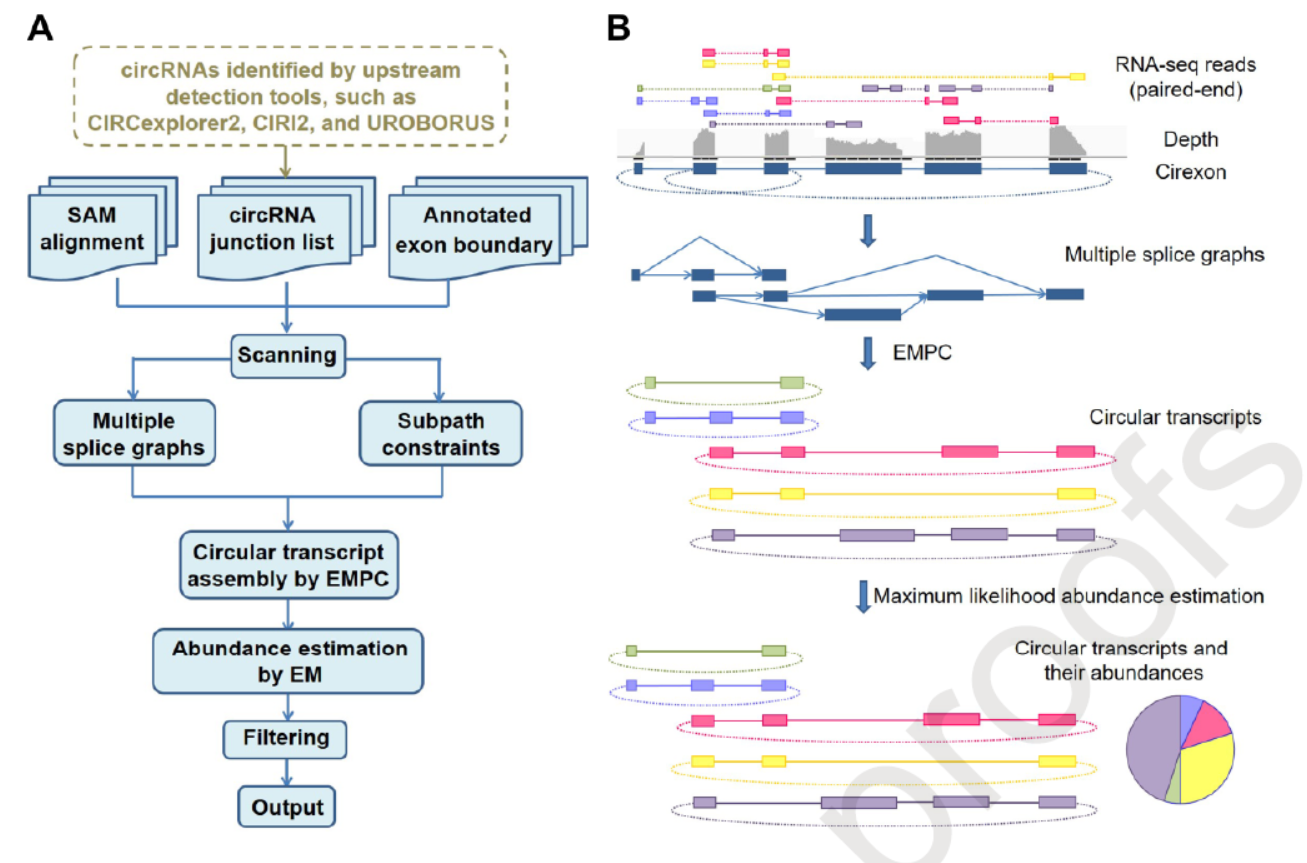
图1 circAST工作原理
circAST分析circRNA全长的效果怎么样?
为测试circAST是否有效,作者将分离的成年小鼠睾丸中circRNA进行测序分析。利用circAST分析得到了3464种circRNA,CIRCexplorer2分析到3892种circRNA分子,两种工具的结果有交叉,也有不同。利用circAST工具,从3464个circRNA中共成功分析获得了2883种circRNA的全长序列和表达丰度信息。作者也进一步对这些circRNA可变剪切方式进行了统计分析和PCR后Sanger测序验证。
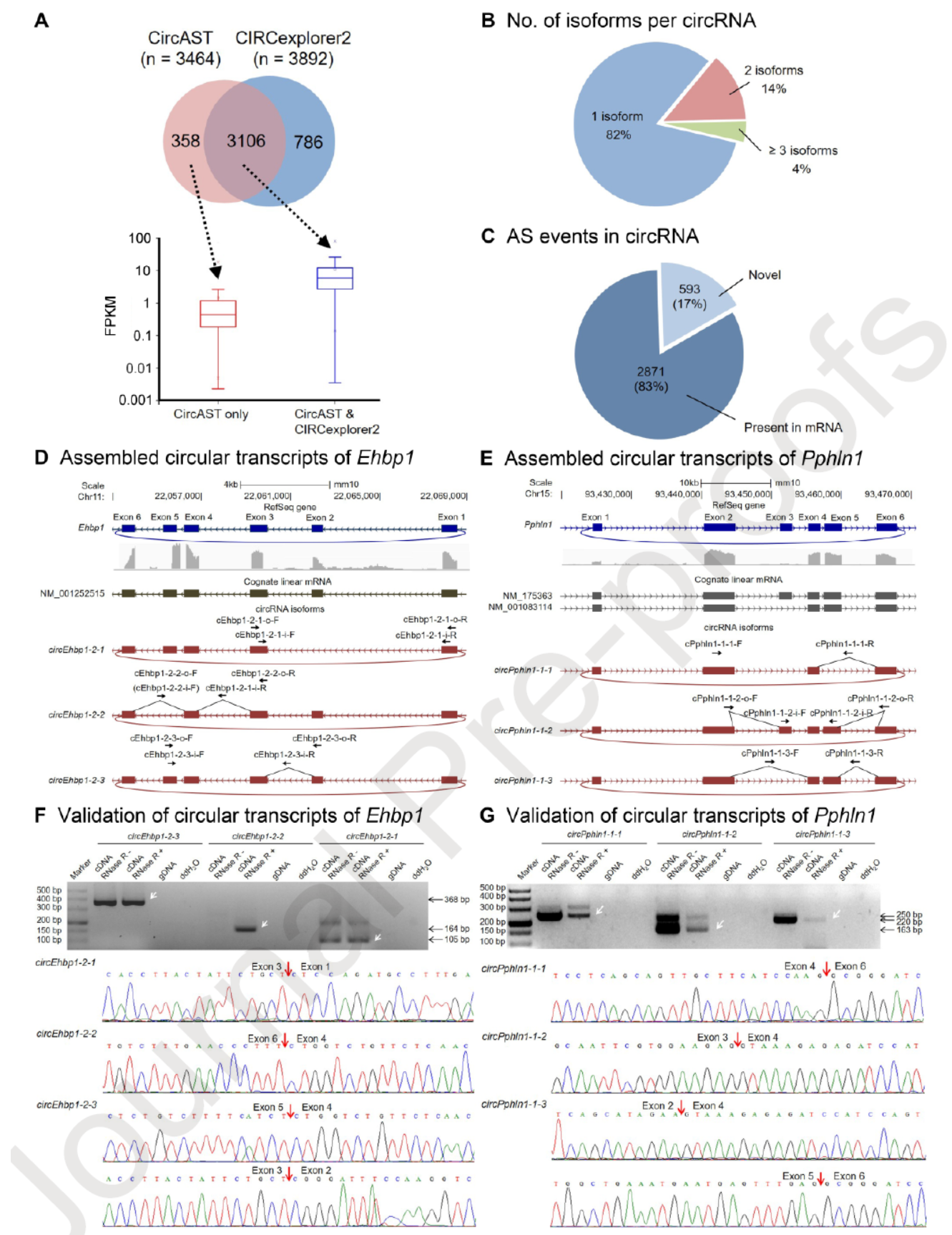
图2 circAST分析成年鼠睾丸circRNA ([1])
circAST与其他circRNA分析工具的比较
circAST分析得出的circRNA丰度信息包括FPKM(fragments per kilobase million mapped reads)和TPM(transcripts per kilobase million mapped)以及circRNA全长所对应的Reads数。由于从分析的一开始就考虑到了所有测到的反向拼接事件,理论上circAST分析circRNA全长序列和丰度的信息可能会更准确。那么实际测试的效果怎样呢?
为分析circAST的效果,作者选择了多种在线数据通过circAST和不同工具进行比较,结果表明,circAST总体灵敏度可达到85.63% – 94.32%,准确性达到81.96% – 87.55%。而CIRCexplorer2的灵敏度(74.75% – 75.61%)和准确性(58.42% – 60.42%)较之低很多。
Sailfish-cir可以对同一反向拼接位点进行精确的丰度分析,但不能识别同一BSJ中不同剪切变体。作者比较分析后结果表明circAST比Sailfish-cir的定量更准确。CIRI-AS是已报道的可以分析circRNA可变剪切的工具,但对同一数据的分析结果显示,circAST能够分析得到更多的可变剪切信息。CIRI-full是另一种已报道的可以分析circRNA可变剪切的工具,通过比较PE100,PE150,PE200和PE250的测序数据进行分析,结果表明对于短读长的测序数据,circAST比CIRI-full的性能更优异,说明circAST更能适应不同测序方案。
circAST是基于外显子注释的方法,但CIRI-AS和CIRI-full不依赖于外显子注释的信息,因此circAST会丢失一些内含子来源或基因间序列构成的circRNA。CIRI-full丢失的circRNA信息往往是全长大于600的circRNA分子。CIRI-full对于读长小于600的测序数据能很好的进行circRNA的分析,并且读长越高结果越好。但circAST在短读长的测序数据分析中有一定的优势。以CIRI-full分析的数据为基准,circAST能从更短读长的测序结果中得到较好的灵敏度,PE100 的可以达到76.05%,而PE150的则可达到80.46%。
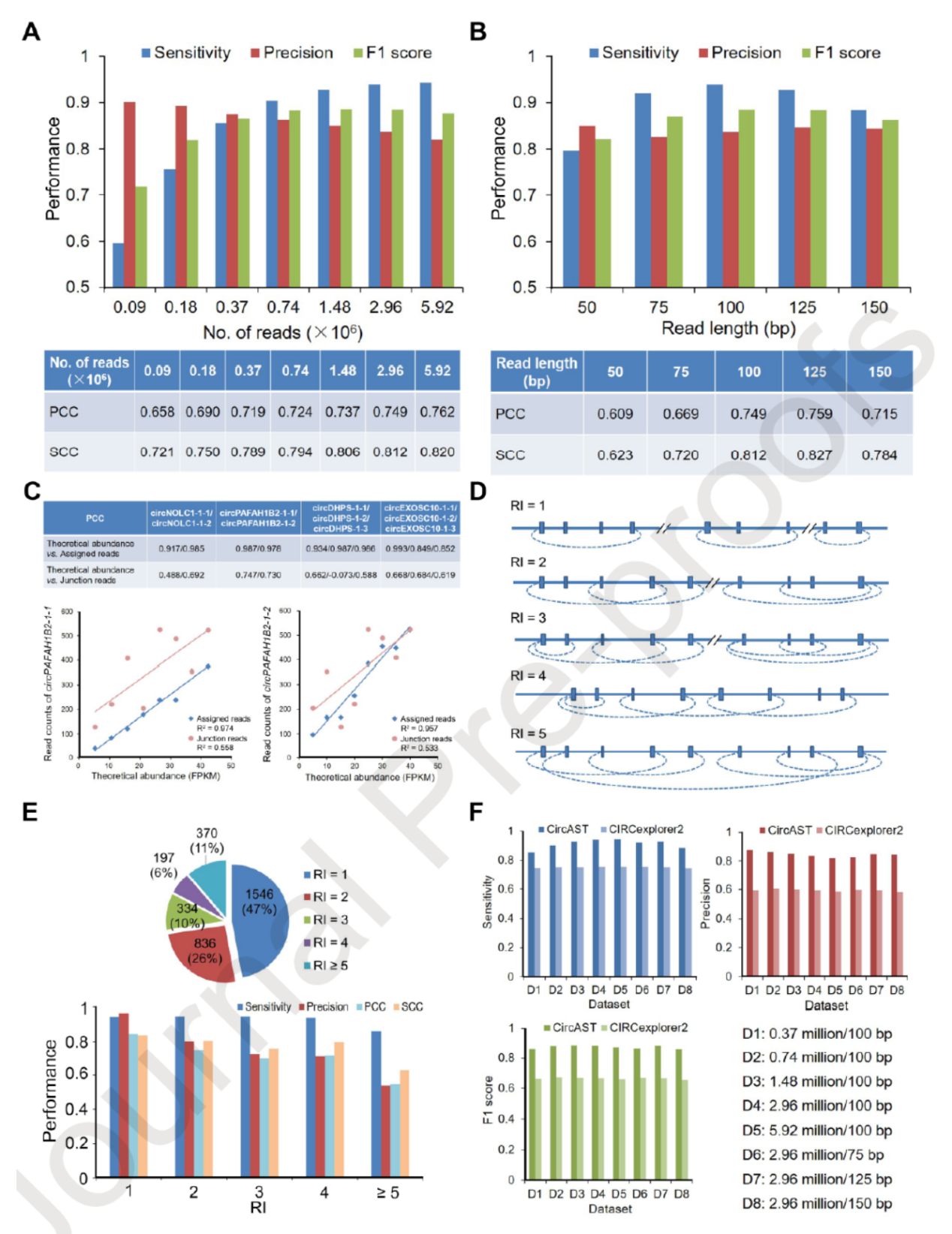
图3 circAST与其他circRNA分析工具的比较分析 ([1])
circAST在短读长的测序数据中依然可以很好地实现circRNA全长序列的分析,这对于大家无疑是个非常好的工具。短读长的测序成本已经非常低,利用这个工具也可以在不用增加测序成本的前提下实现circRNA全长序列的分析,对于那些丰度较高的circRNA分子的分析已经足够了。
最后,circAST是免费开源的工具,下载地址:
https://github.com/xiaofengsong/CircAST
参考文献:
1. Wu, J., et al., CircAST: Full-length Assembly and Quantification of Alternatively Spliced Isoforms in Circular RNAs. Genomics Proteomics Bioinformatics, 2020.