众所周知,circRNA是通过反向剪接位点(BSJ)的非规范剪接产生。大多数现有方法仅通过相应的BSJ识别circRNA,但并不能估计它们的完整序列或识别来自相同BSJ的不同选择性剪接的环状异构体(isoform)。
为此,来自德国医学系统生物学研究所Meyer教授团队专门开发了一个全新的工具——CYCLER,可用于识别新的和选择性剪接的circRNA的完整序列并进行表达定量,同时可估计已知线性RNA剪接异构体的丰度,为circRNA的研究带来了新的便利。该项成果发表在了Nucleic Acids Research(IF=16.971) 期刊上。

1.算法优势
CYCLER的优势之一是它在插入片段大小或RNA-seq输入文库的读取长度方面没有任何隐含或明确的限制。此外,与其他工具相比,CYCLER中基因组特征的量化也不像其他工具那样强烈依赖于高测序深度,因为这种方法仅依赖于junction reads的量化。因此,CYCLER可以利用整个RNA-seq文库进行转录本组装,而不仅仅是恰好跨越剪接位点的大约20%的reads的一小部分。
第二个显著优势是CYCLER能够利用BSJ识别工具的组合。CYCLER的主要优势是装配算法,但也不能完全归功于算法。性能上的差异很大程度上归因于CYCLER更好的基因组特征选择,这有助于减少错误重建的数量。与此相反,CIRI-full仅限于组装短的 circRNA。CIRCexplorer2在组装方面表现不佳,因为它使用了一种从头线性组装的工具——Cufflinks/Stringtie。由于这些工具不能很好地处理circRNA案例,这样就容易引发错误,然后在管道中传播。CIRCexplorer2的另一个缺点是过度依赖注释来检测AS事件。
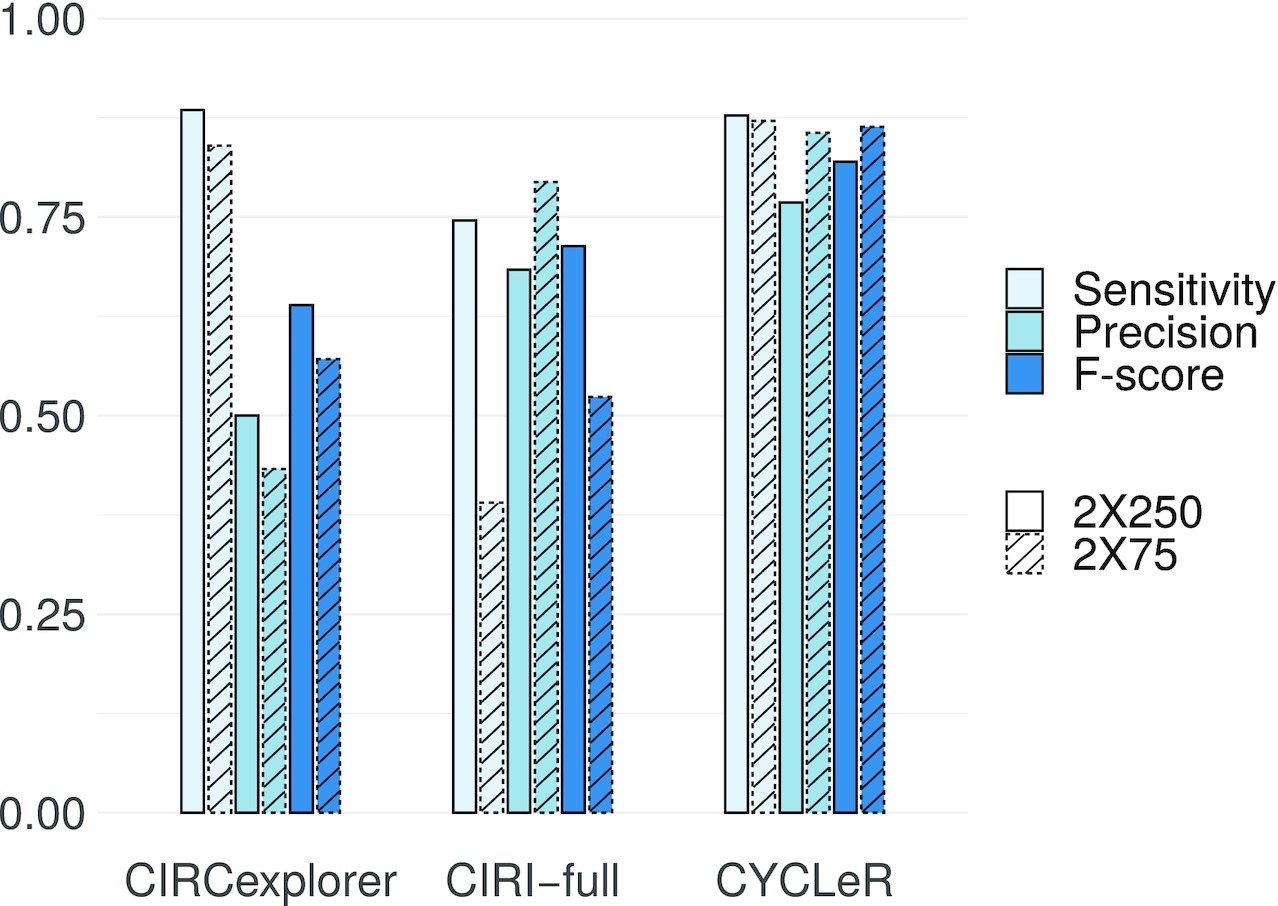
图1.CYCLER和类似工具的比较基准测试
该结果仅基于两种工具正确识别的转录本数量,通过这种方式来判断这些程序的输出以及组装精度对转录本量化的影响。CYCLER是目前唯一一种同时量化已知线性和新组装的环状转录本的方法。
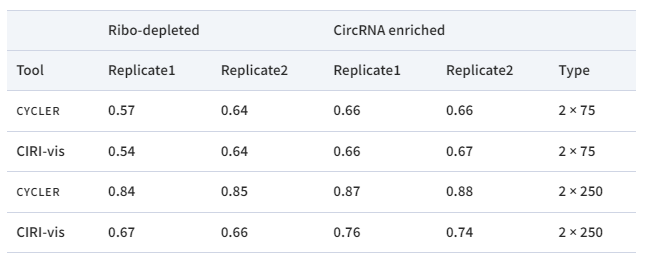
表1. 预测与模拟的circRNA转录本计数的相关性
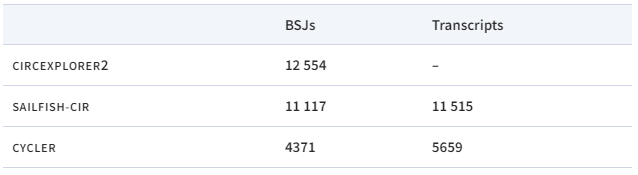
表2. 黑腹果蝇数据集:已识别转录本的总数
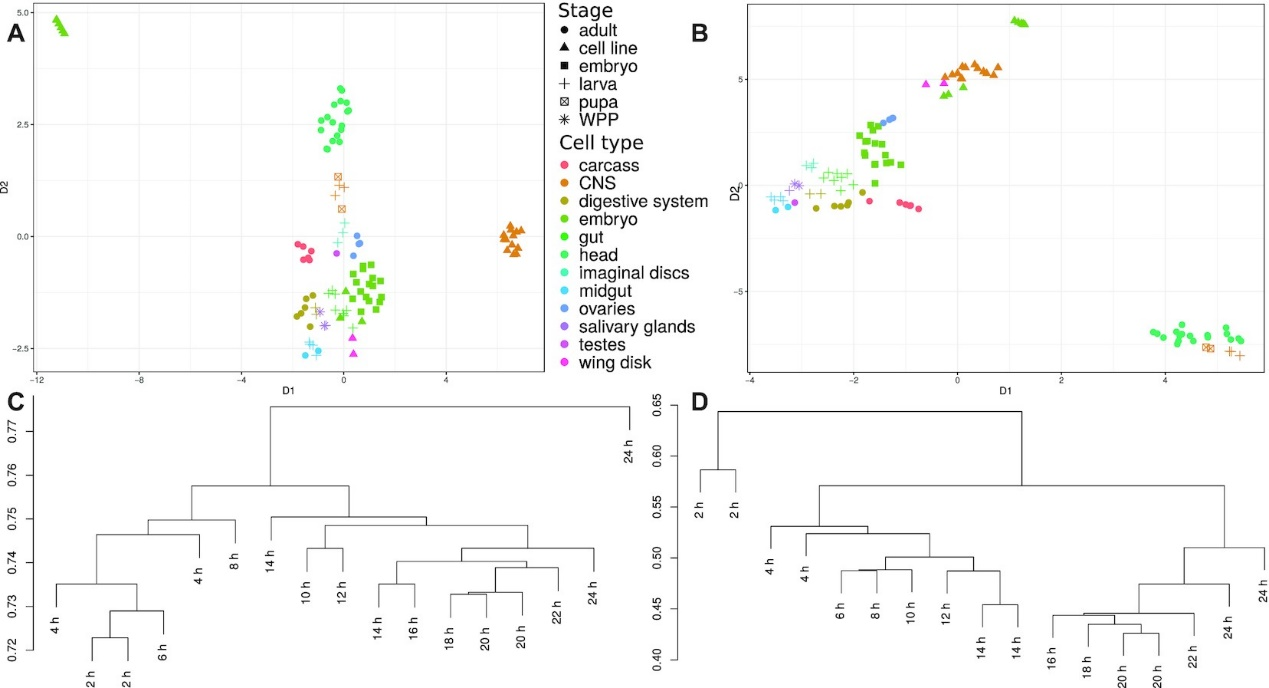
图2. 比较CIRCexplorer2和CYCLER的黑腹果蝇转录组集
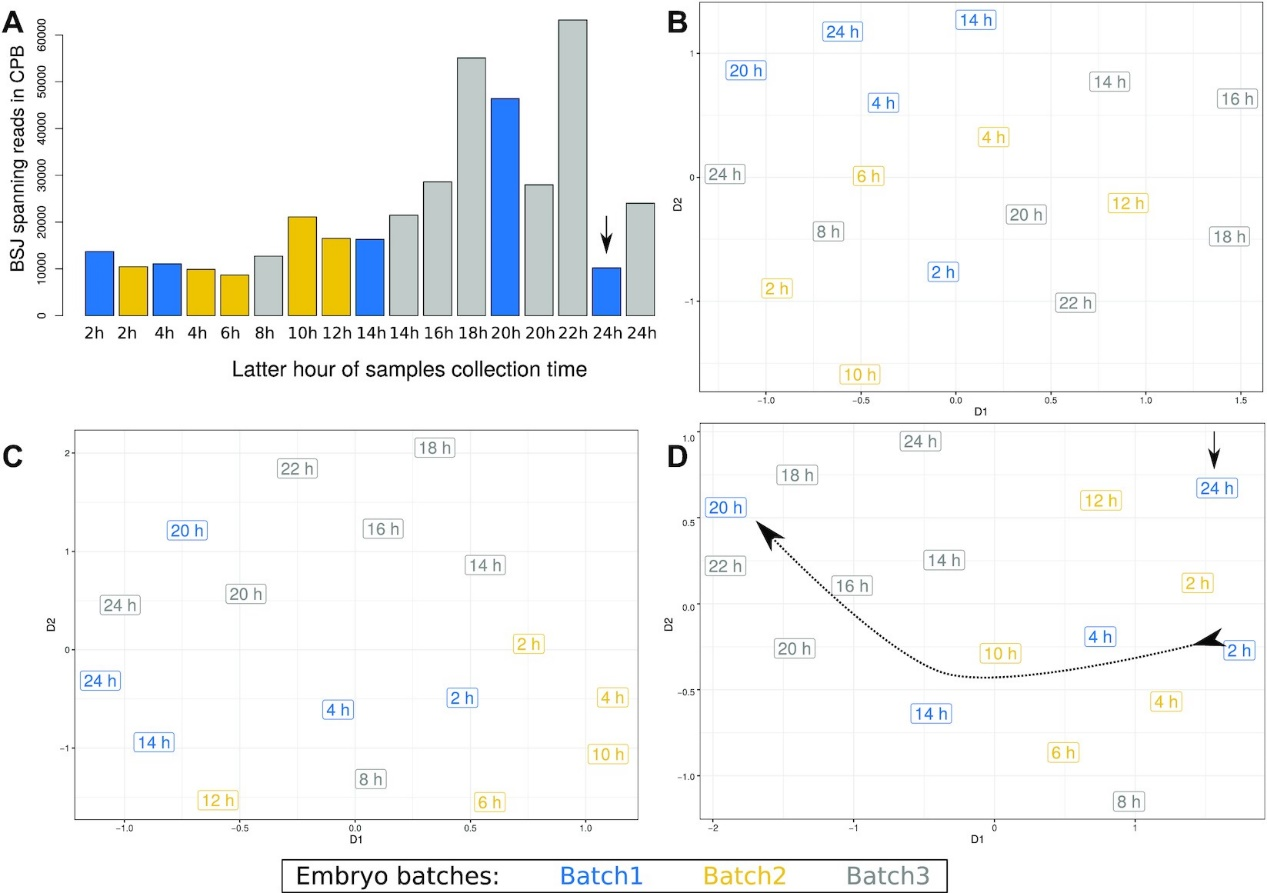
图3. Lai 2014数据集中的批处理效应
作者观察到,与其他基于Illumina的工具相比,CIRCexplorer2 输出的isoform数量非常多。CIRCexplorer2确实提供了最多数量的和CIRI-long共有的isoform;然而,来自CIRCexplorer2的未经CIRI-long验证的isoform数量也高得不成比例。
CYCLER比CIRI-full具有更多数量的和CIRI-long共有的isoform以及更多数量的未经CIRI-long验证的isoform。作者观察到CYCLER比CIRI-full有更多的和CIRI-long共有的结果。然而,这两种工具都具有类似的约 60% 未经CIRI-long验证isoform的比例。这表明在调整序列长度时,CYCLER和CIRI-full的精度相当。CYCLER报告的isoform数量有所增加,而独有的BSJ数量与CIRI-full相似。这可以通过更长的circRNA更可能具有替代isoform这一事实来解释。因此,与CIRI-full相比,CYCLER预测的isoform数量增加是由于CYCLER能够正确组装甚至长剪接isoform。
总之,CYCLER和CIRCexplorer2在验证的转录本方面都优于CIRI-full,而CIRCexplorer2中的误报数量使得该工具不适合isoform组装。CIRI-full受isoform长度的限制——即使有合理的精度统计,该工具也忽略了大量和CIRI-long共有的案例。总而言之,在分析长读长数据方面,CYCLER再次成为唯一在精确率和召回率之间取得良好平衡的工具。
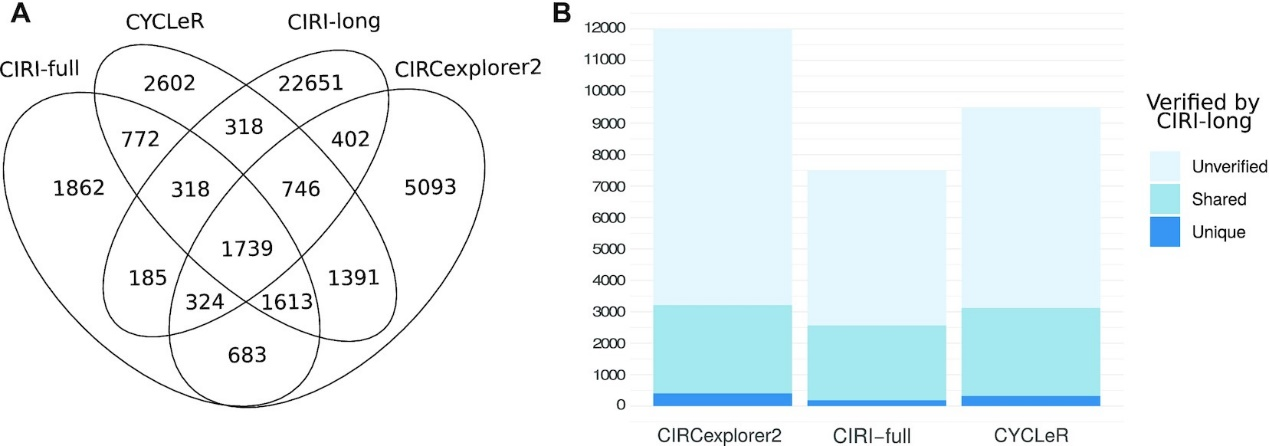
图4. 与CIRI-long数据的对比研究
02.方法策略
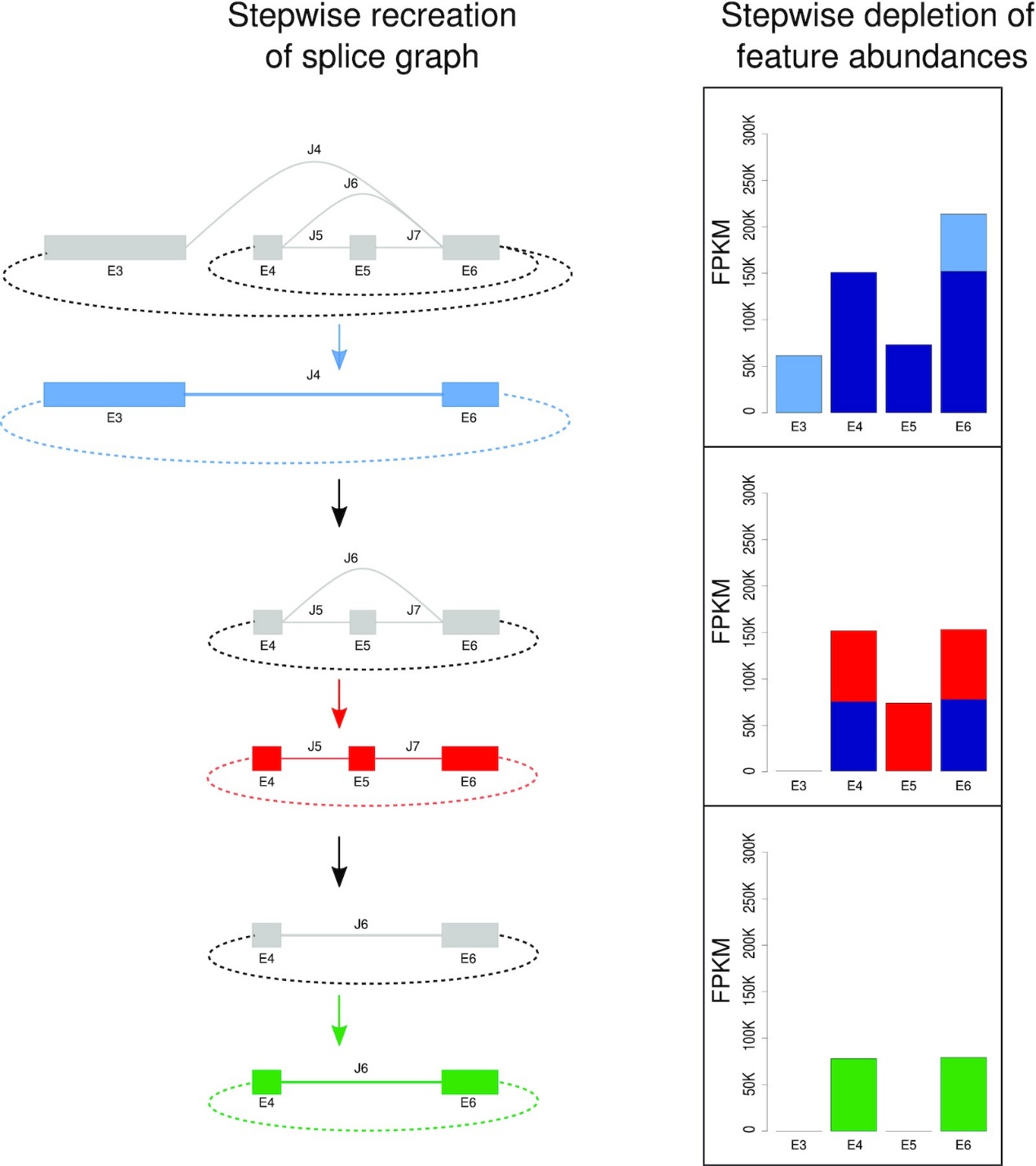
图5.以5-HT2A基因为例在CYCLER中重建环状转录本
03.小结
CYCLER在模拟数据的所有方面都优于现有的circRNA识别和定量工具。同时,使用CYCLER进行转录组分析所需的实验工作量大大低于现有方法。
Stefanov, Im M .CYCLER- a novel tool for the fullisoformassembly and quantification of circRNAs[J]. Nucleic Acids Research, 2022.
转载请联系邮箱授权:circRNA@163.com