1、目前circRNA的命名方式有哪些?
CircRNA自发现以来,数量在不断增加,光circBase数据库收录的人circRNAs数量就达到14万多条,还有很多circRNAs并未收录如circbase。已发表的文献来看,circRNA命名也时各种都有,熟悉circRNA的命名类型在阅读文献时将带来不少方便。
Circbase数据库中circRNA的命名方式主要采用阿拉伯数字来表示,比如hsa_circ_0000284(circbase数据库ID号),has表示物种位人,circ表示circRNA分子,0000284为唯一标示的数字码。同时circbase还给出了这个circRNA的名称hsa_circ_000016。
显然这种阿拉伯数字命名方式给理解circRNA分子比较费劲,所以很多文献直接用circHIPK3来指代这个分子,但实际上HIPK3宿主基因位置可以产生circRNAs多大20个(circbase收录)。circBank数据http://www.circbank.cn/则综合考虑两种需求,采用宿主基因名加数字的方式较好地解决了这个问题,circBank数据库用hsa_circHIPK3_004(hsa_circ_0000284),从circbank的命名方式中即可以获知circRNA来源宿主基因,又可以知道该宿主基因可能产生多个circRNAs。
另外一些circRNA的基因芯片探针则用circRNA通过下划线加阿拉伯数字的方式呈现,如circRNA_013779,circRNA_008008, circRNA_003724,如果直接用这个名称在circbase中检索是查不到对应的相关信息的。
CIRCpedia数据库也有对应的circRNA命名ID系统,如:HSA_CIRCpedia_63389,HSA_CIRCpedia_63393。
2、高通量测序和基因芯片的区别?
高通量测序(RNA-seq)相比于微阵列基因芯片(microarry)主要有以下区别。
- RNA-seq是开放系统: RNA-seq不同于基因芯片,检测基因转录本不需要依赖已知基因组或转录组的参考序列,RNA-seq可以通过比对或拼接的方法,分别检测有参考序列和无参考序列的转录组。基因芯片一个重要的缺点,它是一个封闭的系统,只能检测已知的序列或有限的变异,而RNA-seq的最大优势,它是一个开放的系统,能发现和寻找新的信息。
- RNA-seq动态范围大: RNA-seq最低可以检测(即灵敏度)到总RNA中千万分子一的表达量,只要足够的测序深度,最高表达量不受限制,而芯片由于非特异性杂交带来的噪声,不能检测低丰度表达的转录本,而且,超过一定丰度,检测会产生饱和现象。如Affymetrix芯片上最多检测50000个拷贝,超过这个数值,检测信号不会增大,另外,芯片的非特异性杂交还带来的背景噪声还影响了检测准确度。
- 信息丰富: RNA-seq提供了更为丰富的序列信息,包括可变剪切、融合基因或SNP等大量序列变异信息,而且链特异性技术可以测定转录本来自DNA哪条链。
- 可重复性: 许多因素降低了芯片实验的可重复性,造成了同一类芯片样本之间的相似度大幅降低,而RNA-seq实验可重复性非常高,同类样本间的相关系数往往能够达到0.9以上。
3、转录组测序每组设多少个重复样本比较合适?
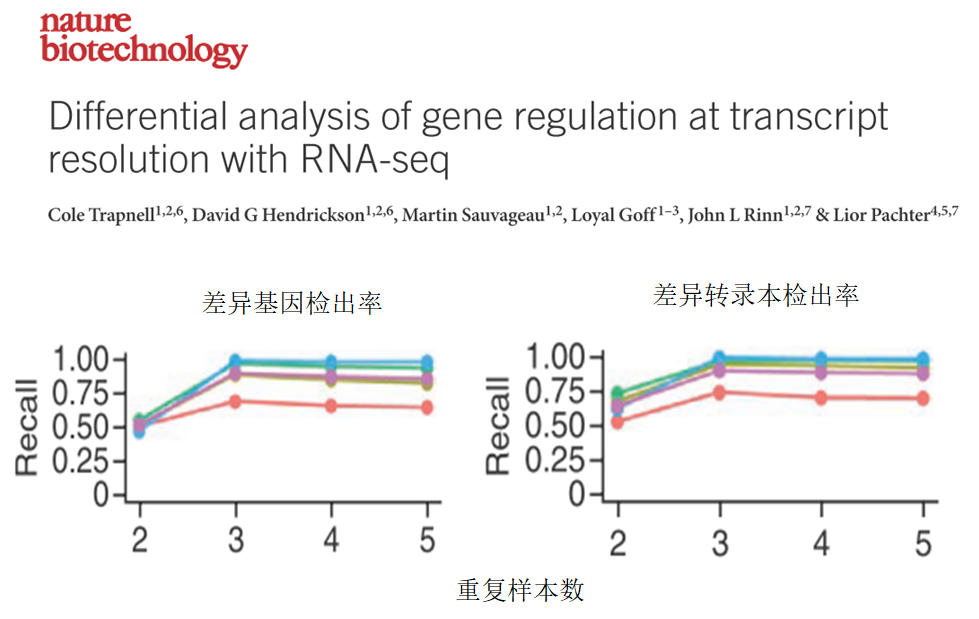
生物学重复指的就是样本重复,比如3只同类型小鼠,在同样的条件下进行处理,即为三个生物学重复。生物学重复对于测序实验数据的解读和分析非常重要。生物体往往存在较大的异质性,个体差异大,设置生物学重复可减少组内误差,降低背景差异,增强结果的可靠性,还可以检测到离群样本,一些异常样本的存在严重影响测序结果的准确性,通过样本间主成分分析可以发现异常样本,可以在后续分析中排除。当生物学重复样本设置到3时,才能得到较为可靠的差异表达基因。Nature Biotechnology一篇文献专门探讨了这个问题,结论是在RNA-seq实验时,设置3个以上的样本重复非常必要,结论见下图。对于一些异质性高的临床样本,这个重复数应设置的更高。
4、高通量测序序列文件FASTQ和FASTA文件格式有何区别?
FASTQ和FASTA文件是存储测序序列(reads或DNA片段)的常用格式,是后续序列比对,序列组装或进化树构建的基础源数据。FASTQ格式由4行组成,以@开头,FASTA格式由2行组成,以 > 开头。FASTQ格式储存的信息更多一些。
举个栗子!For FASTA格式
这个序列是从circBank数据库下载的

- 第一行开头的 ”>” 用于序列标记。hsa_circFLT3_015是序列在circBank中唯一的ID号。
FASTA是数据库中储存序列的一种格式,不适合储存下机的测序数据。因为它没有序列的质量信息。那有测序质量信息的FASTQ格式就成了储存测序数据的常用格式啦!
再来一个栗子!for FASTQ
下面是Illumina平台测序的真实数据,其中包含了1条reads的信息。
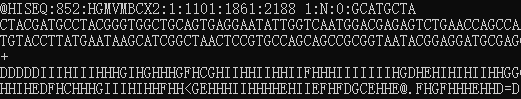
- 第1行主要储存序列测序时的坐标等信息:
@ 序列标记符号
HISEQ:852:HGMVMBCX2 测序仪唯一的设备名称
- lane的编号
- tail的坐标
- 在tail中的X坐标
- 在tail中的Y坐标
1:N:0:GCATGCTA reads1上的INDEX
- 第2行 序列信息,一般用ATCGN表示,其中N表示无法判断的碱基。
- 第3行以 “+” 开头,可以储存附加信息,一般为空
- 第4行 质量信息,与第2行的序列相对应
5、FastQC质量报告中重点关注的内容是哪些?
测序数据分析中通常用FastQC软件对reads进行质量评估,FastQC的结果可以重点关注以下几个方面。
Basic Statistics:对测序数据量、长度和GC含量基本统计;
Per base sequence quality:reads每个位置测序质量;
Per sequence quality scores:每条序列的测序质量分布;
Per base sequence content:统计reads每个位置ATCG四种碱基的分布;
6、如何判断测序得到的reasds序列碱基质量?
测序reads中每个碱基质量如何,主要体现在Per base sequence quality图中,如下图
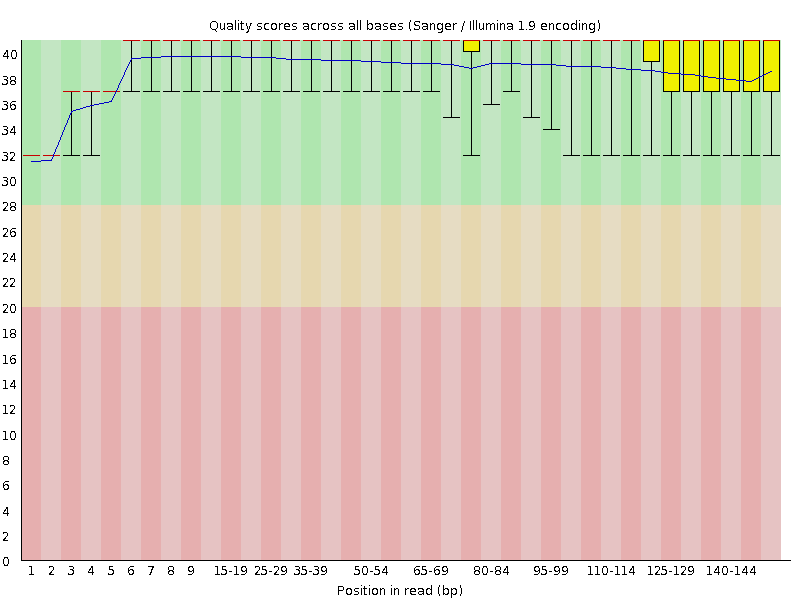
上图横坐标代表每个每个碱基的位置,反映了读长信息,比如测序的读长为150bp,横坐标就是1到150;纵坐标代表碱基质量分数值。图中的箱线图代表在每个位置上所有碱基的质量值分布,中间的红线代表的是中位数。用黄色填充的区域的上下两端分别代表上四分位数和下四分位数;箱线图最上方的短线代表90%,最下方的短线代表10%;蓝色的线代表平均值。背景色从上到在下依次为green, orange, red; 分别代表very good, reasonable, poor;将碱基质量分成3个不同的标准。当有一个位置的10%四分位数小于10或者中位数小于25时会给出警告;当有一个位置的10%四分位数小于5或者中位数小于20时会提示失败。
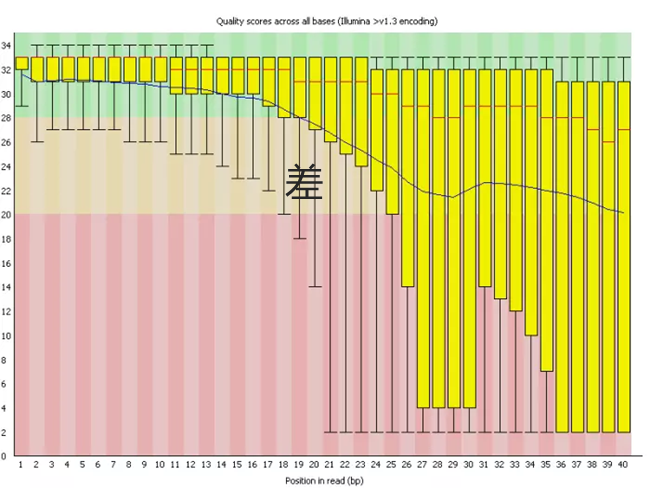
当序列质量差时,将得到如下图:
7、测序样品主成分分析(PCA)用来干嘛,有什么意义?
主成分分析(PCA)是一种数据降维技巧,它能将大量相关变量转化为一组很少的不相关变量,这些无关变量称为主成分。主成分分析应用非常广泛,一次转录组高通量测序分析会获得成千上万的基因表达值,显然很难通过这么多基因表达值直接看出样本间异同,通过主成分分析就可以降低基因维度,直观查看样本间基因表达异同。
因为基因间存在相互调控关系,这些互作的基因间存在表达量相关性,PCA主成分析可以将样本中成千上万个基因表达量维度信息降维到主要几个相关性较高的基因集,这样就可以方便地进行样本间比较,并实现最大程度地保留原始数据信息和代表样本特征,考察样本的变异情况。下图是正常和疾病两组样本的mRNA测序样本间主成分分析的例子。
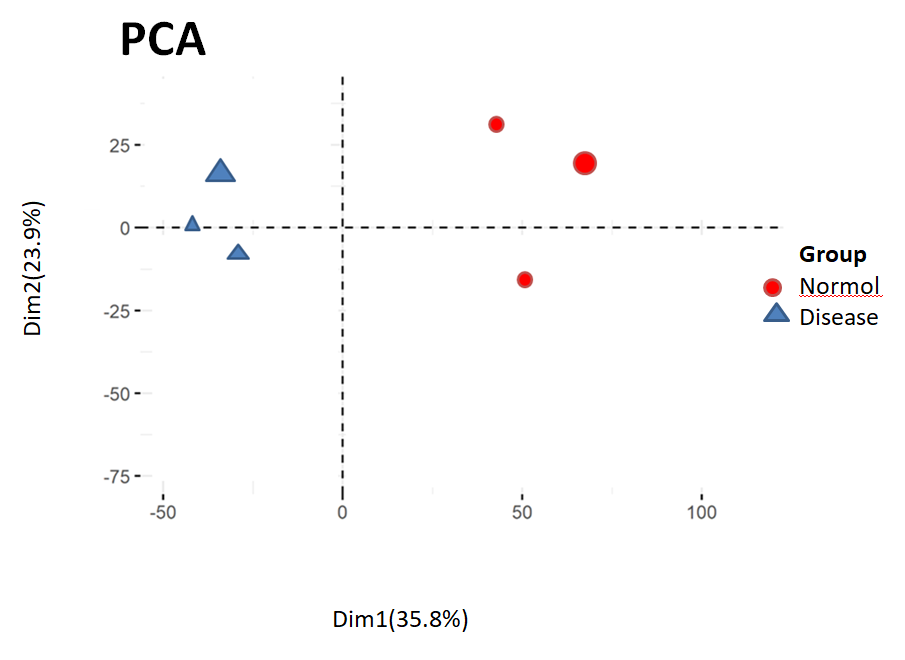
Dim1表示第一主成分,Dim2表示第二主成分,Dim1可解释原所有变量(所有基因表达量)总体方差的35.8%,Dim2可解释原所有变量(所有基因表达量)总体方差的23.9%,Dim1和2两个维度解释总体方差的59.7%。统计学语言解释可能还是不够直白。从图上的各组点聚集情况可以看出,同一组的样品往往会聚类在一起,组间的样品会分隔开,异常样品往往会和其他组内样品分隔开, 如果检测到异常样品,在差异分析时,该样品应该被排除在外。
8、差异表达基因的FDR有何意义,它和p-value有什么关系?
测序完成后,往往能得到上百或上千个差异表达基因,对每个差异基因进行扩大样本qPCR验证似乎不太现实,通常会选取差异倍数越大,p值或FDR值越小的基因进行优先验证。但p值和FDR值究竟有什么统计学意义呢,它们间又有什么联系呢?
假如通过差异比较分析发现,某个基因A在两组样本间差异p-value小于0.05,我们知道任何一种测量手段都可能存在误差,那么基因A是存在真实差异还是测量误差,p-value值小于0.05的意思就是基因A不存在差异的概率小于0.05,换言之测量的随机误差小于0.05,但这个判断还是有0.05的犯错概率,就里就是假阳性率(False positive rate),但这只是一次判断,FDR值计算过程则是对p-value的多次判断校正即多重检验,降低假阳性率。RNA-seq分析中普遍采用BH(Benjamini and Hochberg)多重检验校正法,通过FDR法可以得到每个基因p-value校正后的q-value,通常FDR、Q value、Adjusted p-value是指同一个东西。FDR值比p-value更严格,数值越小越可靠,但没有约定的阈值,不像p-value小于0.05和0.01时才认为差异显著和差异非常显著。
9、IPA数据库的优势主要体现在哪里?
RNA-seq实验获得差异表达基因后,通常根据GO和KEGG免费数据库进行基因功能富集分析,这些免费数据库往往存在更新不及时,缺乏人工审校的缺点。IPA (Ingenuity PathwayAnaylsis)数据分析系统则可以弥补上述缺点,对差异基因实现更可靠的分析。IPA中各个分子互作,功能注释模块都由专家进行编译,来源于文献,是非常可靠的生物学大规模关系型数据库,全面涵盖了蛋白质、基因、复合物、细胞、组织、药物、通路和疾病信息,收录信息达600万条,并且每周实时更新,是分析基因功能的一把利器,目前使用IPA处理数据发表文献超过2万篇。IPA不仅可以将目标基因进功能富集分类,还可以预测上下游调控关系,并根据下游基因表达状态预测上游调控因子是被激活还是被抑制,分析结果中用p-value表示富集显著性,Z-score表示激活或抑制效应,阈值一般为2和-2。下图是IPA通路分析常见图型。
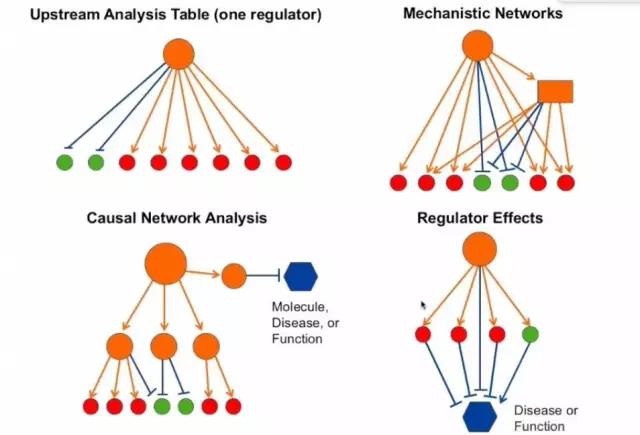
RegulatorEffect是综合多个分析模块结果的一个分子调控图,通过该预测图,可推导出哪些调控因子如何通过下游靶分子直接或间接导致疾病的发生。不难看出,这种预测结果为指导后续实验提供了非常有价值的线索。
10、差异基因跟qPCR验证结果不一致如何办?
转录组测序后得到差异基因后,一般都需要进一步进行qPCR验证,可能面临qPCR结果与测序结果不一致的情况。从技术上来说,qPCR更为准确,但测序通量更高,方便用来进行前期基因筛查。两个技术平台,很难做到100%的一致性差异。比如验证30个基因,有25个表达趋势一致,另外5个基因如果PCR结果有差异,则以PCR结果为准。不一致的情况下,我们重点要排除以下几个方面的原因。
- 实验组和对照组是否设置颠倒;
- 保证检测样本的一致性,意思是用同样的测序备份样本或RNA进行PCR验证;
- 应重点选择高表达的基因,验证时选择低表达的基因比例过高时,容易出现不一致情况;
- 挑选差异基因时,是否只看RNAseq中的p-value,FDR值是否太高(生物学重复少少时应提高FDR阈值,不能只看p-value);
- 检查qPCR实验中内参基因Ct值是否稳定,排除PCR的实验问题;
- 通过测序PCA结果,在qPCR实验中排除异常样本;