RNA与蛋白相互作用是RNA功能研究的焦点问题之一,也是环状RNA研究者接下来经常会遇到的问题。本文将通过解读有关方法学资料介绍一种RNA-蛋白相互作用的验证实验:电泳迁移率改变(EMSA)实验。主要参考2011年发表在 Methods in Molecular Biology上的一篇方法学文章(Maxwell, 2011) 。

电泳迁移率改变实验(Electrophoretic Mobility Shift Assay,EMSA)是常用的研究核酸-蛋白相互作用的技术,基本原理是如果核酸分子与蛋白存在相互作用,电泳中会因为两者结合而导致电泳速度变慢,通过杂交实验可以看出具体的变化状态。该实验的操作相对简单,还能进行定量分析,是研究RNA-蛋白相互作用非常有用的技术。
材料与试剂:
常规材料:
重蒸酚(Tris-HCl, pH 8.0);
氯仿:异戊醇(24:1);
RNase-Free超纯水;
3M乙酸钠(pH 5.2);
无水乙醇;
70%乙醇
放射标记RNA制备用试剂材料:
小牛肠磷酸酶(CIP)及10×CIP buffer:0.5 M Tris-HCl (pH 9.0), 100 mM MgCl2, 10 mM ZnCl2, 0.1 M spermidine-HCl
多核苷酸激酶(PNK)及10×PNK buffer:0.5 M Tris-HCl (pH 7.6), 70 mM MgCl2, 50 mM 二硫苏糖醇(DTT)
[γ-32P] 标记ATP
G-25 sephadex 凝胶珠与离心柱 (Amersham Pharmacia)
TE buffer:10 mM Tris-HCl (pH 7.5), 1 mM EDTA.
丙烯酰胺母液:acrylamide:bisacrylamide=19:1
10×TBE:0.89 M Tris base, 0.89 M 硼酸, 20 mM EDTA
尿素(分子生物学级)
10% 过硫酸铵(APS):现用现配
TEMED
Gel loading buffer: 80%甲酰胺, 1×TBE, 10 mM EDTA
溴酚蓝和二甲苯蓝染料
Clear plastic wrap (SaranTM wrap)
Black India ink
RNA洗脱缓冲液:0.3 M乙酸钠, 5 mM EDTA, 10 mM Tris-HCl, pH 7.4, 0.1% SDS
磷屏或X-胶片
0.45-μm孔径的针头滤器
EMSA试剂:
Buffer D: 20 mM HEPES (pH 7.0), 0.1 M NaCl, 3 mM MgCl2, 0.4 mM EDTA, 1 mM DTT, 20% 甘油
10×binding buffer:0.1 M HEPES, pH 7.0, 1 M NaCl
10×phosphate dye: 25 mM 磷酸钾 (pH 7.0), 25%蔗糖, 0.1 mg/mL 溴酚蓝
10×phosphate buffer: 0.25 M 磷酸钾 (pH 7.0)
甘油(分子生物学级)
3mm厚度Whatman滤纸
实验方法:
RNase-Free技术要求:
- 玻璃器皿通过高温消毒方法实现RNase-Free,DEPC处理塑料制品;
- 实验全程带手套,避免皮肤来源的RNase污染;
- 避免重复使用枪头;
- 保持操作台面整洁无尘,移液枪等设备定期清洗;
- 所有试剂保证无RNase污染;
- RNA样品操作要格外小心,尽量低温操作。避免RNA样品接触二价离子,高pH值(>9.0)或长时间高温放置。
酚-氯仿提取RNA:
- 向样品中加入等体积的苯酚(注意事项1),剧烈震荡混匀;
- 10000×g 离心 3 min上层水相转移至新的RNase Free的EP管中(注意事项2);
- 重新向苯酚中加入等体积的RNase-Free水混匀后离心收集水相;
- 向两次收集的水相中加入等体积的氯仿,剧烈震荡混匀,10000×g 离心 3 min;
- 上层水相转移至新的离心管中,加入1/10体积的3 M 乙酸钠(pH 5.2),加入预冷的无水乙醇至终体积比浓度为70%,–20℃ 放置大于 1 h(注意事项3);
- 室温>10000×g 离心20 min ,小心去除上清;
- 用等体积的预冷70%乙醇洗涤沉淀,轻轻颠倒混匀数次,立即室温>10000×g 离心20 min ,小心去除上清;
- 冻干法或阴凉处晾干,沉淀用RNase-Free水溶解,测量浓度,保存备用(注意事项4)。
放射标记RNA制备:
常见的放射标记的RNA分为全标记和末端标记两种方式。本方法主要介绍末端标记的探针制备与分离纯化的方法(注意事项5)。
- 探针分子去磷酸基:
在1.5mL的RNase-Free EP管中按下列体积配制反应体系:
RNA探针 20μg
10×CIP buffer 20μL
CIP (1U/μL) 10μL
ddH2O 补充至总体积为200μL
37℃孵育45min。酚-氯仿法纯化RNA样品。沉淀用30μL RNase-Free水溶解,测量浓度,保存备用。
- 探针末端标记:
在1.5mL的RNase-Free EP管中安下列体积配制反应体系:
CIP-treated RNA (1–2μg) 50–80 pmol
10×PNK buffer 2.5μL
[γ-32P] ATP (1μCi/μL) 8–10μL
PNK (20 U/μL) 1μL
ddH2O 补充至总体积为25μL
- 37℃孵育1.5h。添加25 μLddH2O,酚-氯仿法纯化RNA样品。沉淀用30μL RNase-Free水溶解,测量浓度,保存备用。
为有效去除未准确标记至探针分子中游离[γ-32P] ATP,需要进行探针纯化。探针纯化的方案有两种:酚-氯仿抽提结合分子筛纯化的方法;基于凝胶纯化的方法。凝胶纯化法能获得更高纯度的探针样品。
酚-氯仿抽提结合分子筛纯化:
- 将待纯化的探针体系加至2-cm bed of G-25 分子筛凝胶柱(Amersham Pharmacia)上(注意事项6),<1,000×g离心3 min;
- 用闪烁计数器检查Cerenkov计数的放射性(注意事项7)。注意不要使用闪烁液。将1μL的洗脱液放入闪烁小瓶中的0.5mL微量离心管中进行计数。
- 标记好日期和放射性计数并将纯化后的探针储存在-20ºC。 注意32P的半衰期为14.2天。
凝胶纯化法纯化:凝胶纯化法能获得纯度更高的探针,尤其是存在样品降解的情况。
- 准备40mL 6% 丙烯酰胺溶液 (丙烯酰胺:甲叉双丙烯酰胺=19:1),1×TBE 和7 M 尿素溶液(注意事项8);
- 加入10% APS (8μL/mL) 和TEMED (1μL/mL),颠倒混匀,灌胶,室温聚合20min;
- 用1×TBE冲洗并预电泳20min,电流控制在40–45 mA(注意事项9);
- 向待纯化探针体系中加入Loading Buffer,样品煮沸3-5min,室温冷却后上样电泳,电流控制在40–45 mA,电泳时间大约1-2h;
- 将电泳胶板撬开后用塑料膜覆盖暴露的一侧,用混有放射性染料(1μL[γ-32P] ATP)的30μL黑色印度油墨标记凝胶的三个角,风干凝胶;
- 用干净的胶带盖住干燥的墨滴,并在上面放上一个phosphorimager卡带。暴露5-10分钟(注意事项10)。在phosphorimager中扫描磁带,并在玻璃板下面打印出凝胶。对齐点并用热处理的剃须刀片切出放射性RNA带;
- 用1mL的Tip头将凝胶片捣碎,加入500μL的RNA洗脱缓冲液,室温下摇动45分钟(注意事项11);
- 10000×g离心2分钟回收洗脱的RNA。通过0.45μm注射器过滤器将洗脱液(凝胶头顶部的溶液)过滤到新的1.5 mL EP管中;
- 用300μL的RNA洗脱缓冲液重复洗脱。再次混匀并通过相同的注射器过滤以与之前的洗脱液合并。将洗脱液分成两个管(每个400μL)和乙醇沉淀物(不添加乙酸钠)。
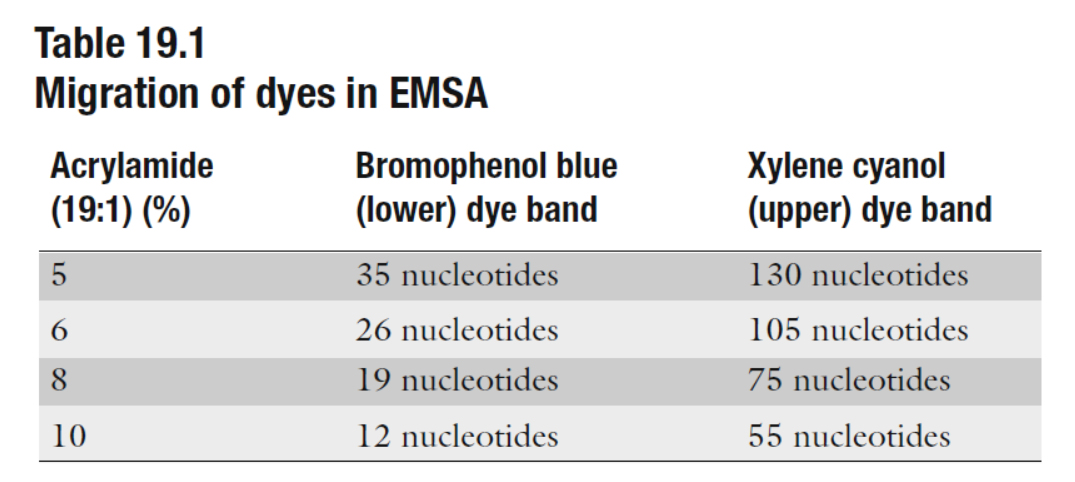
表1 不同浓度凝胶分离效果 (来自(Maxwell, 2011))
EMSA实验检测RNA-蛋白相互作用:
RNA结合蛋白结合反应体系:
- 按下图的体系配制反应体系:
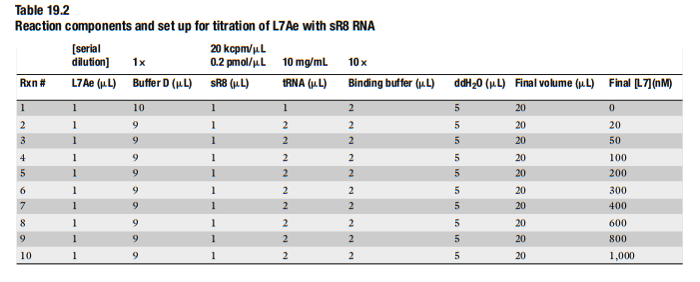
表2 RNA结合蛋白结合反应体系 (来自(Maxwell, 2011))
将10×binding buffer与tRNA加水混匀(注意事项12);
加入sR8 RNA(注意事项13);
加入Buffer D 和L7Ae 蛋白(注意事项14)。
- 轻轻混匀体系,70℃反应8分钟(注意事项15)。自然冷却至室温,离心弃沉淀,上清转移至新的离心管中,加入2μL 10×phosphate dye,轻轻混匀。
Native非变形电泳胶制备:
- 准备40mL 6% 丙烯酰胺溶液 (丙烯酰胺:甲叉双丙烯酰胺=19:1),1×phosphate buffer和2%甘油(注意事项16);
- 加入10% APS (8μL/mL) 和TEMED (1μL/mL),颠倒混匀,灌胶 (尺寸16×18×1.5 cm),插入15孔梳子(注意事项17),室温聚合20min;安装玻璃板,加入1×phosphate buffer作为电泳液(注意事项18);
RNA-蛋白复合物电泳分离:
- 尽量采用冷循环电泳液的体系进行电泳(注意事项19),尽量降低电泳体系的发热造成的影响,(文中建议的是Hoefer SE600);
- 150V电泳,约1.5~2h,溴酚蓝电泳至凝胶2/3位置。0
电泳结果成像:
- 分开玻璃板,使凝胶粘在一块板上;
- 将两片比凝胶略大的3mm Whatman滤纸切下。 将滤纸压在凝胶上。 从纸上剥下纸(注意事项20)。 凝胶将粘在纸上;
- 用透明塑料膜包住凝胶的暴露面,并在80℃的真空下在凝胶干燥器中干燥1小时。
- 干燥时,丢弃滤纸背面(吸收多余的液体和放射性物质)。将凝胶暴露于phosphorimager盒过夜或X射线胶片2-4小时。
注意事项:
- 使用苯酚时要特别注意。理想的情况下,苯酚应该重新蒸馏。使用前在0.1M Tris-HCl缓冲液中平衡(pH8.0)。添加0.2%的β-巯基乙醇可防止氧化并延长保质期。在-20℃存储重蒸和平衡苯酚,4℃储存短期内用的。-20℃放置6月后或者如果变色(最常见的是粉红色),则应丢弃。
- 酚和水层有可能会变成相反的分层,例如使用非常高盐或蔗糖浓度的溶液。在提取之前或之后加入少量的水区分哪一层是含水的。小心不要收集含水相的苯酚。为了获得最佳结果,将一些水相留在苯酚顶部以确保不收集苯酚。 如果用水相收集苯酚,则将其在氯仿萃取步骤中除去。 在RNA沉淀过程中,苯酚/氯仿提取的最后一步中所携带的氯仿除去。
- 非常稀的RNA溶液通常不会完全沉淀。在这里介绍的方法一般不会出现这个问题。 但是,如果出现了,可在-20℃孵育过夜(16小时)提高沉淀效率。此外,可以添加少量的辅助剂来帮助沉淀,例如糖原或后期反应中安全的非特异性RNA(经常使用来自大肠杆菌或酵母的tRNA)。
- 使用比尔定律从260nm处的吸光度(如果消光系数是已知的)计算RNA浓度,或者将在260nm处的OD值乘以40μg/ OD / mL的一般转换因子来计算RNA。 这可得到以μg/ mL为单位的RNA浓度值。
- 如果所使用的RNA是合成的,那么它没有5’磷酸,这一步应该省略。 如果RNA在体外转录或从提取物中纯化,则需要去磷酸化。
- 使用前,G-25树脂应在TE缓冲液中溶胀并平衡。摆动式转子将提供更清洁的分离和更有效的去除游离[γ-32P] ATP。
- 切伦科夫计数不要使用闪烁液。将1μL洗脱液放入闪烁小瓶中的0.5mL微量离心管中进行计数。
- 对于特定RNA,可以根据要分辨的RNA的大小选择凝胶百分比(参见表1)。 我们在实验室中用1×TBE,7M尿素保存20%丙烯酰胺的原液,并用1×TBE,7M尿素的原液稀释,得到所需的最终丙烯酰胺百分比。 这样可减少每次新鲜的准备工作,这是不必要的和耗时的。室温下避光保存,1个月后丢弃。
- 凝胶预电泳是必要的,能提高凝胶的温度,手触感觉非常温暖或稍微有点热的比较理想。凝胶中的热量和尿素有助于保持RNA变性。一般而言,电泳RNA直到其通过凝胶的方式移动大约2/3(参见表1)。电泳过程需要注意避免凝胶过热。在电泳过程中,玻璃板应该是温热的,但不要发烫,如果板子太热有可能会开裂。 如果最终的电泳结果呈现出“微笑”样,中间比外部样品跑得快,则表明凝胶加热不均匀。 应该通过减少通过凝胶的电流来避免太多“微笑”样的带型。
- 尽量不要调整phosphorimager图片的大小,如果有必要可调整对比,然后打印。也可将凝胶在暗房里用X光胶片曝光5-15分钟,显影后的胶片可以放在玻璃胶板的下面。
- 此处的洗脱步骤可以在4℃摇动过夜完成。
- L7Ae的储存和稀释缓冲液是缓冲液D。要直接将蛋白质溶液直接加入反应中,而不是直接加到反应管的两侧。
- 在这些反应中,tRNA在超过摩尔浓度时用作非特异性RNA。其存在确保只观察到特定的RNA结合,并在凝胶上提供更好的RNP分辨率。
- 放射性标记的RNA应与非放射性标记的RNA混合,制成每立方厘米20,000 cpm和0.2 pmol RNA的储备液。每次反应20,000cpm一般而言是足够的,但也可以优化显影条件。
- M. jannaschii是L7Ae蛋白与sR8 box的高亲和力高效结合C/D sRNA需要高温。 大多数RNA结合蛋白最佳结合的范围从4到37℃。常常加入过量的非特异性蛋白如牛血清白蛋白(BSA)以保持蛋白质水平处于相对恒定的浓度,防止蛋白质沉淀,并提供更清楚的RNP解析。由于在用于结合的极端热条件下可能发生的聚集和沉淀,BSA不在这些反应中添加。 最终反应物含有20mM HEPES,pH 7.0, 0.15M NaCl,0.5mM DTT,0.2mM EDTA,10%甘油,1.5mM MgCl2,1mg / mL tRNA。
- 甘油是凝胶的重要组成部分,不要忽略。在成像显影之前干燥凝胶期间,甘油防止凝胶收缩和破裂,这可能使凝胶不能用于出版。 甘油也被认为有助于RNP的解决。
- 在灌胶之前应彻底清洗玻璃板,并保持不含有清洁剂。玻璃板可以在组装之前用乙醇擦拭或冲洗以除去灰尘。玻璃板也可以烘烤,虽然彻底的清洗往往足以去除污染的RNA酶。
- 1×phosphate buffer对真菌和细菌生长敏感,应在使用前从10×储备液中新鲜制备。 如果在电泳期间或之后在顶部和底部之间重新循环,运行缓冲液可重复使用多次,但最好每次现配现用。
- 点样时要小心,尽量避免将样品与电泳缓冲液混合。首先将点样器的头插到点样孔的底部,然后慢慢点样。当快要加完的时候先把Tip头提起,慢慢取走。避免从Tip头吹出气泡以避免样品被带出或稀释。Native凝胶不具有压缩的效果,因此点样的过程要尽量使样品紧,这对形成清晰的条带非常重要。凝胶必须保持冷却以获得最佳分辨率。冷水自来水通常是足够的。不要让凝胶的温度升高超过室温几度。
- 如果凝胶浓度较高(> 14%),凝胶可能不会粘在滤纸上。 在这种情况下,将保鲜膜置于凝胶上,翻转凝胶并铺平,然后将凝胶剥离。 凝胶应粘在保鲜膜上。 然后可以将滤纸放在凝胶的顶部以进行干燥。 对于低浓度凝胶(4%),凝胶可能非常容易变形,使得凝胶带在干燥和显影之后波状起伏。 将凝胶从平板转移到滤纸时要小心。 如果凝胶不会粘附在一块板上,那么将ddH2O喷到凝胶上会有所帮助。
参考文献:
Maxwell, K.T.G.a.E.S. (2011). Electrophoretic Mobility Shift Assay for Characterizing RNA–Protein Interaction. Methods in molecular biology.